Object splitting for in-silico mixed scRNA-seq data and Slide-Seq data¶
This example uses TACCO to annotate in-silico mixtures of mouse colon scRNA-seq data and mouse colon slide-Seq data (Avraham-Davidi et al.) and split them into “pure” measurements corresponding to single cell type only.
(Avraham-Davidi et al.): Avraham-Davidi I, Mages S, Klughammer J, et al. Integrative single cell and spatial transcriptomics of colorectal cancer reveals multicellular functional units that support tumor progression. doi: https://doi.org/10.1101/2022.10.02.508492
[1]:
import os
import sys
import matplotlib
import pandas as pd
import numpy as np
import anndata as ad
import tacco as tc
# The notebook expects to be executed either in the workflow directory or in the repository root folder...
sys.path.insert(1, os.path.abspath('workflow' if os.path.exists('workflow/common_code.py') else '..'))
import common_code
Load data¶
[2]:
reference_data_path = common_code.find_path('results/slideseq_mouse_colon/data')
plot_path = common_code.find_path('results/object_splitting', create_if_not_existent=True)
[3]:
reference = ad.read(f'{reference_data_path}/scrnaseq.h5ad')
tdata = ad.read(f'{reference_data_path}/slideseq.h5ad')
Plotting options¶
[4]:
highres = False
default_dpi = 100.0
if highres:
matplotlib.rcParams['figure.dpi'] = 648.0
hr_ext = '_hd'
else:
matplotlib.rcParams['figure.dpi'] = default_dpi
hr_ext = ''
axsize = np.array([3,3])*1.0
labels_colors = pd.Series({'Epi': (0.00784313725490196, 0.24313725490196078, 1.0), 'B': (0.10196078431372549, 0.788235294117647, 0.2196078431372549), 'TNK': (1.0, 0.48627450980392156, 0.0), 'Mono': (0.5490196078431373, 0.03137254901960784, 0.0), 'Mac': (0.9098039215686274, 0.0, 0.043137254901960784), 'Gran': (0.34901960784313724, 0.11764705882352941, 0.44313725490196076), 'Mast': (0.23529411764705882, 0.23529411764705882, 0.23529411764705882), 'Endo': (0.8549019607843137, 0.5450980392156862, 0.7647058823529411), 'Fibro': (0.6235294117647059, 0.2823529411764706, 0.0)})
Define splitting tasks¶
Generate a reference with balanced type composition, i.e. with the same number of cells for each type
[5]:
n_labels = reference.obs['labels'].value_counts()
n_labels = n_labels[n_labels>200]
min_n = n_labels.min()
balanced = reference[np.concatenate([ tc.utils.complete_choice(reference.obs.query(f'labels == {label!r}').index, min_n) for label in n_labels.index ])].copy()
Generate in-silico mixtures of scRNA-seq data to benchmark methods with a known ground truth
[6]:
def mixit(reference,capture_rate):
tdata = tc.tl.mix_in_silico(reference, type_key='labels', n_samples=len(reference), bead_shape='gauss', bead_size=1.0, capture_rate=capture_rate,)
tdata.obsm['reads_labels'] /= tdata.obsm['reads_labels'].to_numpy().sum(axis=1)[:,None]
return tdata
Define the tasks as reference data + mixed data
[7]:
problems = {
'balanced reference\n capture_rate=1.0': [balanced,mixit(balanced,capture_rate=1.0),],
'balanced reference\n capture_rate=0.1': [balanced,mixit(balanced,capture_rate=0.1),],
' full reference\ncapture_rate=1.0': [reference,mixit(reference,capture_rate=1.0),],
' full reference\ncapture_rate=0.1': [reference,mixit(reference,capture_rate=0.1),],
'full reference\n slide_seq': [reference,tdata,],
}
Create comparison grid¶
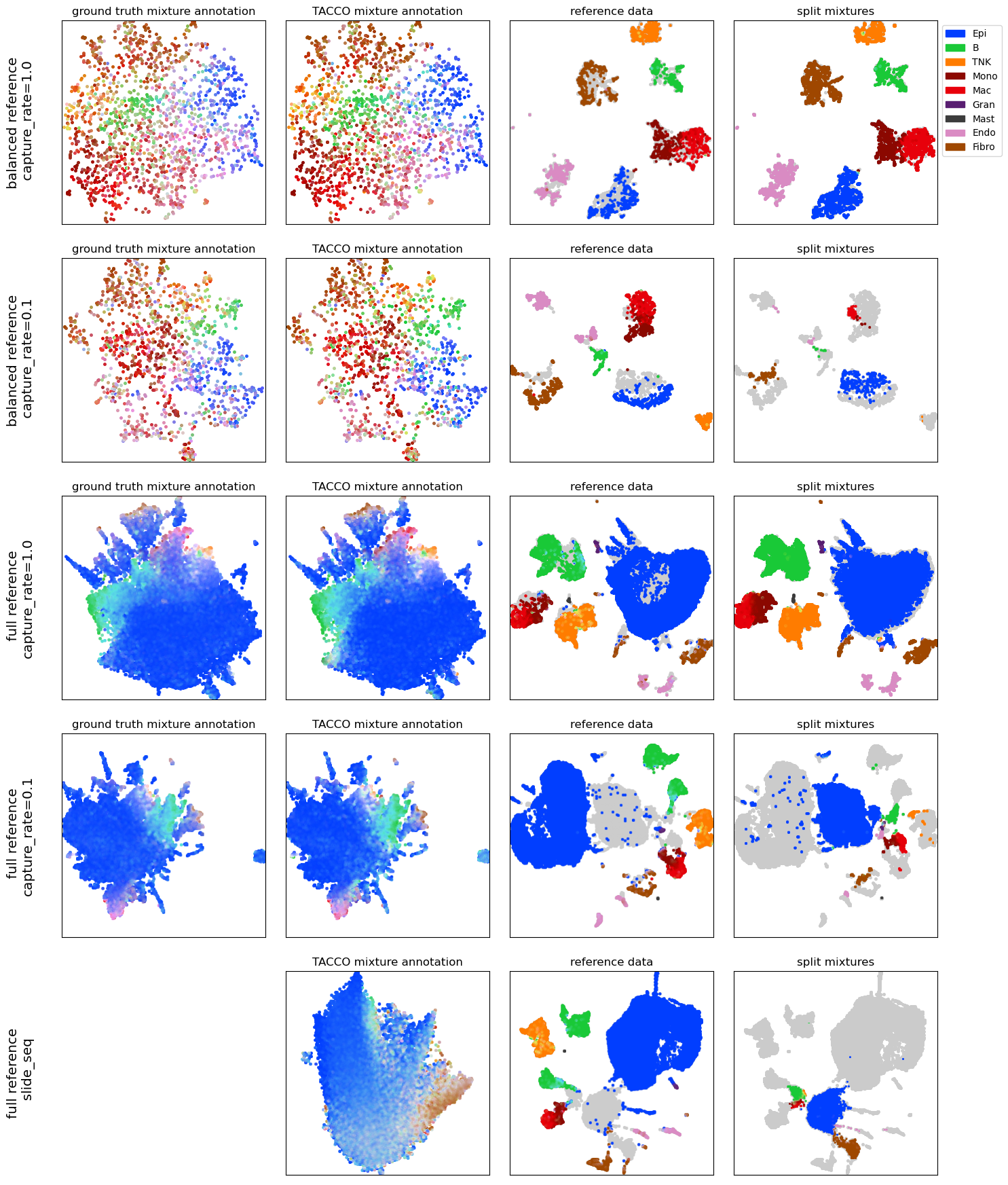
Create grid of plots with tasks in the rows and UMAPs showing different aspects in the columns:
ground truth mixture annotations (where available)
TACCO mixture annotation (generated from the mixtures and the reference)
reference data
split mixtures
[8]:
fig,axs=tc.pl.subplots(4,len(problems),x_padding=0.3, y_padding=0.5,axsize=axsize);
for ip, pn in enumerate(problems.keys()):
ref, adata = problems[pn]
# annotate the mixtures, and keep the extra information for the object splitting
adata, ref2 = tc.tl.annotate(adata, ref, 'labels', result_key='TACCO', multi_center=10, reconstruction_key='rec', return_reference=True)
ref2, adata, ref = tc.pp.filter([ref2, adata, ref], return_view=False, )
# UMAP the mixtures
adata_umap = tc.utils.umap_single_cell_data(adata, hvg=True)
if 'reads_labels' in adata.obsm:
tc.pl.scatter(adata_umap, joint=True, position_key='X_umap',ax=axs[ip,0],legend=False, point_size=10, keys='reads_labels', colors=labels_colors, noticks=True);
axs[ip,0].set_title('ground truth mixture annotation')
else:
axs[ip,0].axis('off')
axs[ip,0].text(-0.2, 0.5, pn, fontsize='x-large', rotation=90, horizontalalignment='center', verticalalignment='center', transform=axs[ip,0].transAxes)
tc.pl.scatter(adata_umap, joint=True, position_key='X_umap',ax=axs[ip,1],legend=False, point_size=10, keys='TACCO', colors=labels_colors, noticks=True);
axs[ip,1].set_title('TACCO mixture annotation')
# split the mixtures
sdata = tc.tl.split_observations(adata, 'rec', map_obs_keys=True, result_key='labels')
# jointly preprocess and umap the split mixtures and the reference
prep_ref = tc.utils.preprocess_single_cell_data(ref, hvg=True)
ref_hvg = ref[:,sdata.var.index.intersection(prep_ref.var.index)]
ref_hvg = ref_hvg[tc.sum(ref_hvg.X,axis=1)>=100].copy()
sdata_hvg = sdata[:,sdata.var.index.intersection(prep_ref.var.index)]
if pn.endswith('capture_rate=1.0'):
thr=100
else:
thr=30
sdata_hvg = sdata_hvg[tc.sum(sdata_hvg.X,axis=1)>=thr].copy()
scat_hvg = ref_hvg.concatenate([sdata_hvg],index_unique=None)
umap_scat_hvg = tc.utils.umap_single_cell_data(scat_hvg, hvg=False)
ref_hvg.obsm['X_umap'] = pd.DataFrame(umap_scat_hvg.obsm['X_umap'],index=umap_scat_hvg.obs.index).reindex(ref_hvg.obs.index)
sdata_hvg.obsm['X_umap'] = pd.DataFrame(umap_scat_hvg.obsm['X_umap'],index=umap_scat_hvg.obs.index).reindex(sdata_hvg.obs.index)
dummy_key = tc.utils.find_unused_key(umap_scat_hvg.obs)
umap_scat_hvg.obs[dummy_key] = 'dummy'
only_gray = {t:'#CCCCCC' for t in umap_scat_hvg.obs[dummy_key].unique()}
tc.pl.scatter(umap_scat_hvg, joint=True, position_key='X_umap',ax=axs[ip,2], legend=False, point_size=10, keys=dummy_key, colors=only_gray, noticks=True)
tc.pl.scatter(umap_scat_hvg, joint=True, position_key='X_umap',ax=axs[ip,3], legend=False, point_size=10, keys=dummy_key, colors=only_gray, noticks=True)
tc.pl.scatter(umap_scat_hvg[ref_hvg.obs.index.intersection(umap_scat_hvg.obs.index)], joint=True, position_key='X_umap',ax=axs[ip,2], legend=False, point_size=8, keys='labels', colors=labels_colors, noticks=True)
tc.pl.scatter(umap_scat_hvg[sdata_hvg.obs.index.intersection(umap_scat_hvg.obs.index)], joint=True, position_key='X_umap',ax=axs[ip,3], legend=ip==0, point_size=10, keys='labels', colors=labels_colors, noticks=True);
axs[ip,2].set_title('reference data')
axs[ip,3].set_title('split mixtures')
Starting preprocessing
Annotation profiles were not found in `reference.varm["labels"]`. Constructing reference profiles with `tacco.preprocessing.construct_reference_profiles` and default arguments...
Finished preprocessing in 0.45 seconds.
Starting annotation of data with shape (1644, 16590) and a reference of shape (1652, 16590) using the following wrapped method:
+- platform normalization: platform_iterations=0, gene_keys=labels, normalize_to=adata
+- multi center: multi_center=10 multi_center_amplitudes=True
+- bisection boost: bisections=4, bisection_divisor=3
+- core: method=OT annotation_prior=None
mean,std( rescaling(gene) ) 1.2639881469699417 0.35777931746825187
bisection run on 1
bisection run on 0.6666666666666667
bisection run on 0.4444444444444444
bisection run on 0.2962962962962963
bisection run on 0.19753086419753085
bisection run on 0.09876543209876543
Finished annotation in 5.97 seconds.
SCumap...SCprep...time 0.8224248886108398
time 26.4715313911438
scale.....time 46.26188349723816
fuseall...time 3.053330421447754
SCprep...time 0.7486720085144043
SCumap...SCprep...time 0.9363586902618408
time 17.342292547225952
Starting preprocessing
Finished preprocessing in 0.35 seconds.
Starting annotation of data with shape (1347, 3326) and a reference of shape (1652, 3326) using the following wrapped method:
+- platform normalization: platform_iterations=0, gene_keys=labels, normalize_to=adata
+- multi center: multi_center=10 multi_center_amplitudes=True
+- bisection boost: bisections=4, bisection_divisor=3
+- core: method=OT annotation_prior=None
mean,std( rescaling(gene) ) 0.03402273053915133 0.04142684087860696
bisection run on 1
bisection run on 0.6666666666666667
bisection run on 0.4444444444444444
bisection run on 0.2962962962962963
bisection run on 0.19753086419753085
bisection run on 0.09876543209876543
Finished annotation in 3.29 seconds.
SCumap...SCprep...time 0.15137195587158203
time 6.57263970375061
scale.....time 11.43474292755127
fuseall...time 0.36908459663391113
SCprep...time 0.31328487396240234
SCumap...SCprep...time 0.10767793655395508
time 7.238934278488159
Starting preprocessing
Annotation profiles were not found in `reference.varm["labels"]`. Constructing reference profiles with `tacco.preprocessing.construct_reference_profiles` and default arguments...
Finished preprocessing in 7.94 seconds.
Starting annotation of data with shape (17464, 20224) and a reference of shape (17512, 20224) using the following wrapped method:
+- platform normalization: platform_iterations=0, gene_keys=labels, normalize_to=adata
+- multi center: multi_center=10 multi_center_amplitudes=True
+- bisection boost: bisections=4, bisection_divisor=3
+- core: method=OT annotation_prior=None
mean,std( rescaling(gene) ) 1.2568713033073806 0.3341021274436307
bisection run on 1
bisection run on 0.6666666666666667
bisection run on 0.4444444444444444
bisection run on 0.2962962962962963
bisection run on 0.19753086419753085
bisection run on 0.09876543209876543
Finished annotation in 85.03 seconds.
SCumap...SCprep...time 9.238901376724243
time 55.995683431625366
scale.....time 184.38863897323608
fuseall...time 128.69577646255493
SCprep...time 7.033684492111206
SCumap...SCprep...time 5.879138231277466
time 69.99655866622925
Starting preprocessing
Finished preprocessing in 7.76 seconds.
Starting annotation of data with shape (15909, 6533) and a reference of shape (17512, 6533) using the following wrapped method:
+- platform normalization: platform_iterations=0, gene_keys=labels, normalize_to=adata
+- multi center: multi_center=10 multi_center_amplitudes=True
+- bisection boost: bisections=4, bisection_divisor=3
+- core: method=OT annotation_prior=None
mean,std( rescaling(gene) ) 0.026516409323954396 0.04025005088115052
bisection run on 1
bisection run on 0.6666666666666667
bisection run on 0.4444444444444444
bisection run on 0.2962962962962963
bisection run on 0.19753086419753085
bisection run on 0.09876543209876543
Finished annotation in 17.6 seconds.
SCumap...SCprep...time 1.2731781005859375
time 28.459827423095703
scale.....time 28.98681902885437
fuseall...time 9.710714101791382
SCprep...time 5.185055255889893
SCumap...SCprep...time 1.9514830112457275
time 42.562610387802124
Starting preprocessing
Finished preprocessing in 15.87 seconds.
Starting annotation of data with shape (33673, 16879) and a reference of shape (17512, 16879) using the following wrapped method:
+- platform normalization: platform_iterations=0, gene_keys=labels, normalize_to=adata
+- multi center: multi_center=10 multi_center_amplitudes=True
+- bisection boost: bisections=4, bisection_divisor=3
+- core: method=OT annotation_prior=None
mean,std( rescaling(gene) ) 0.7410490029315331 10.559549576241444
bisection run on 1
bisection run on 0.6666666666666667
bisection run on 0.4444444444444444
bisection run on 0.2962962962962963
bisection run on 0.19753086419753085
bisection run on 0.09876543209876543
Finished annotation in 22.85 seconds.
SCumap...SCprep...time 16.929930925369263
time 76.88800930976868
scale.....time 22.30505895614624
fuseall...time 7.340550422668457
SCprep...time 11.40554690361023
SCumap...SCprep...time 3.3191449642181396
time 43.770554304122925
[ ]: